For years, when you asked Cheméo to predict a property, the answer came from Joback. Joback is a group-contribution method: you cut the molecule into functional groups, each group carries a tabulated contribution, and you add them up. It is fast and transparent, and on the small, well-behaved molecules it was fitted on, it is good.
It also has one known weakness: the addition itself. The bigger the molecule, the more groups you sum, and the more the small per-group errors pile on top of each other. The first-order groups also miss the longer-range structure a large molecule has. Chemists have patched this by hand for decades, adding second- and third-order groups as corrections, like in the Marrero-Gani method. Those corrections work, but someone has to sit down and define and fit every one of them.
I wanted these corrections to be learned from the data. So I took the experimental measurements sitting in the Cheméo database, one property at a time, and trained a graph neural network on each of them. Those models are live now on chemeo.com/predict, under the name Cheméo Relay. The full write-up, with all the numbers and the failures, is in the preprint.
The idea, and why it should work
A graph neural network reads a molecule the obvious way: atoms are nodes, bonds are edges. It learns a small vector for each atom from the atoms around it, then pools all those atom vectors into a single vector for the whole molecule, and reads the property off that. If the pooling is a sum, the network is doing exactly what a group-contribution method does, adding up local contributions, except the contributions and their corrections are learned end to end instead of tabulated. This was the bet: a learned Joback that does not fall apart on big molecules.
Then six properties came back as noise
I trained one model per property with the framework defaults and looked at the results. Most were fine. Six were a disaster: the critical temperature, the critical pressure, the critical volume, the Gibbs energy of formation, and a couple more all landed at an R² near zero. An R² near zero means the model is no better than a machine that ignores the molecule and always guesses the average of the training set.
The frustrating part is that nothing looked wrong. The training converged, the loss curves went down and flattened out like they should, and at the end the model confidently predicted the average.
So I ran the cheapest sanity check I could think of. I threw a plain gradient-boosted tree, the kind of model you fit in three lines, at the same data, using a dozen basic descriptors. It beat the fancy graph network on all six properties. The data was learnable, the bug was mine.
A flat line at 0.44 L/mol
Here is critical volume, predicted against experimental, drawn twice.
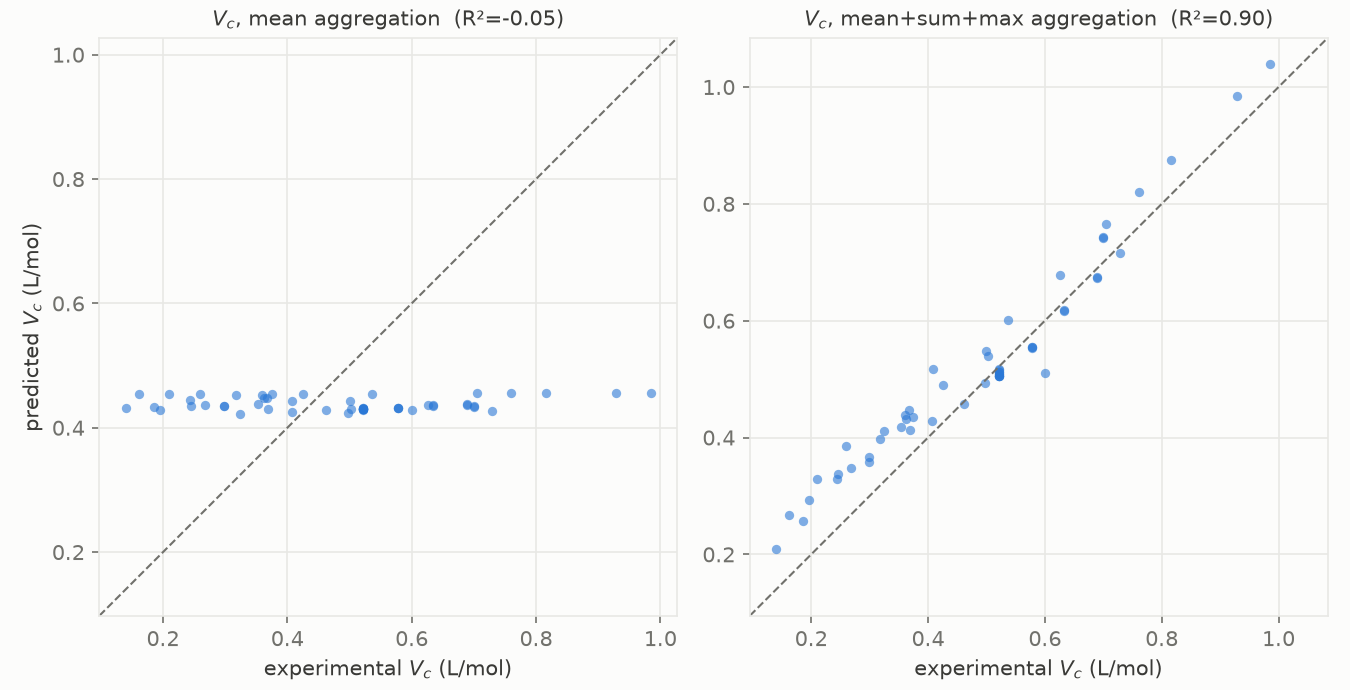
Left, the default. Right, the same network with one thing changed.
On the left, whatever the true critical volume is, the model answers about 0.44 L/mol. It is a flat line. The reason is the pooling step. The default way to combine the atom vectors is to average them, and an average per atom simply throws away how many atoms there are. A molecule and its dimer, twice the size, come out as almost the same vector. For a property like the critical volume, which literally grows with the size of the molecule, the model cannot even represent the thing that changes, so the best it can do is sit on the average.
The fix is one word: replace the average with a sum. Now the molecule vector grows with the number of atoms, the size is back in the model, and the message passing gets to learn the longer-range corrections that Marrero and Gani had to add by hand. Everything else, the network, the data, the split, stayed the same. On the right, it lands on the diagonal. Critical volume went from a flat line to an R² of 0.90, and the formation enthalpy went from an average error of 91 kJ/mol down to 35, just from that one change.
Error against molecule size
The whole point was the big molecules, so the real test is to look at how the error grows with the size of the molecule, not at the average error.
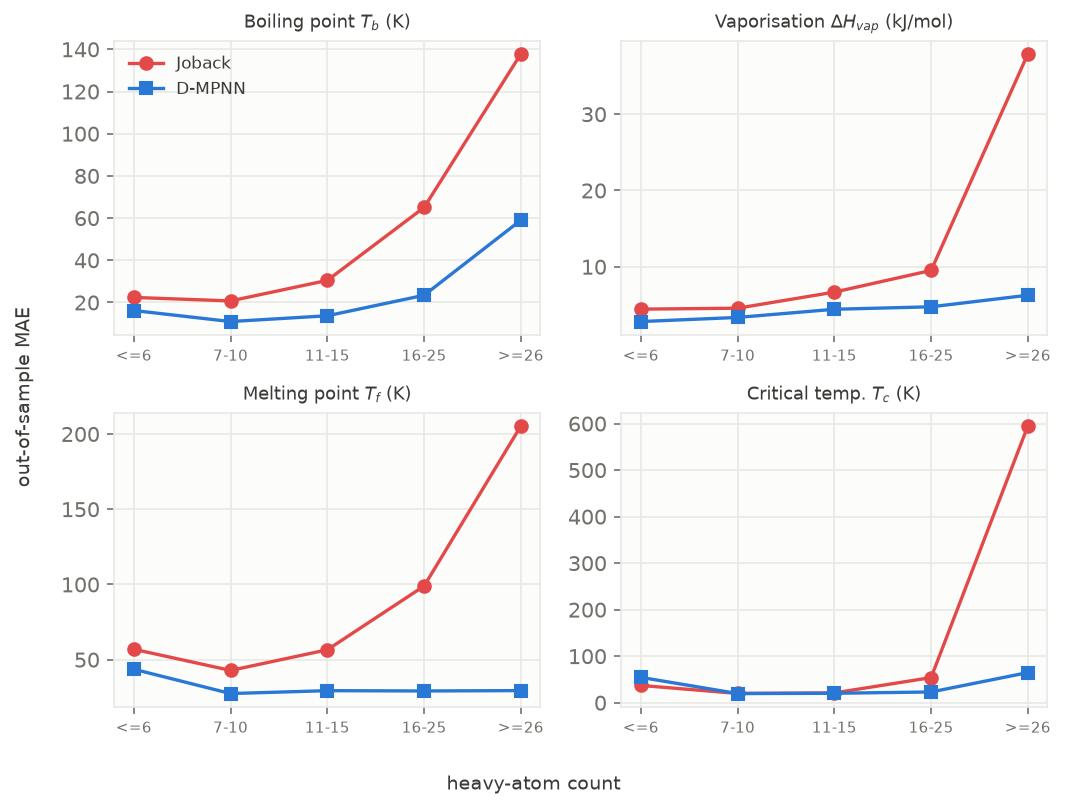
Error against heavy-atom count. Joback in red, Cheméo Relay in blue.
This is the graph I was after. Joback's error climbs steeply with size, exactly what the addition predicts: for the critical temperature it goes from about 37 K on the small molecules to almost 600 K on the largest ones. The neural network stays low and, for the melting point and the critical temperature, essentially flat. On the normal boiling point the numbers are 13.5 K of average error against Joback's 26.3 K, and about 3.5% in relative terms.
What I did not release
I did not release everything, and this is on purpose. Nine properties out of sixteen are live. A property gets released only if it clears a statistical bar and passes a veto: if Joback still predicts it better than my model does, in the range where I can actually measure it, I do not replace Joback, whatever my R² says. Two properties, the Gibbs energy of formation and the critical pressure, are good models on paper but lose that veto, so Cheméo still serves you the Joback value for them and marks mine as beta. Joback has been doing this job for forty years, it does not get replaced by a neural network just because the neural network is new.
You can draw a molecule on /predict and read the numbers straight off. The little editor you draw it in is the under-50-kB tool I finished coding last week, I am pretty happy I got it done before the release of the new models! If you want the rest of the story, the controls, the dead ends, the equations, it is all in the preprint.